- Trang chủ
- Sách y học
- Bài giảng huyết học và truyền máu
- Thalassemia (thiếu hụt chuỗi globin)
Thalassemia (thiếu hụt chuỗi globin)
Bình thường gen α nằm trên cánh ngắn nhiễm sắc thể 16, mỗi nhiễm sắc thể có hai gen α như vậy một cơ thể bình thường có bốn gen α.
Biên tập viên: Trần Tiến Phong
Đánh giá: Trần Trà My, Trần Phương Phương
Định nghĩa
Là bệnh thiếu máu tan máu di truyền gây ra do giảm hoặc mất hẳn sự tổng hợp của một loại chuỗi globin.
Phân loại
Bình thường phân tử huyết sắc tố là α2 β2, có sự cân bằng giữa tổng hợp chuỗi α và chuỗi β. Quá trình tổng hợp một loại chuỗi bị rối loạn sẽ gây thiếu loại chuỗi đó và thừa tương đối chuỗi còn lại làm xuất hiện tình trạng bệnh lý. Nếu tổng hợp thiếu hoặc không tổng hợp được chuỗi α sẽ gây bệnh α thalassemia. Nếu tổng hợp chuỗi β bị hạn chế hay ngừng hẳn sẽ gây bệnh β thalassemia.
α thalassemia
Như đã trình bày ở phần huyết sắc tố, các gen α nằm trên nhiễm sắc thể 16, nếu vùng gen đó tổn thương không tổng hợp được chuỗi gọi là α ° (còn gọi (α1 thalassemia), nếu tổn thương nhưng vẫn tổng hợp được chuỗi a với số lượng ít thì được gọi là α+ (còn gọi α2 thalassemia). Do đó có thể có các kiểu tổ hợp ở người thalassemia.
Đồng hợp tử: α°/ α° Thal.
Đồng hợp tử: α+/ α°+ Thal.
Dị hợp tử α°: α°/α Thal.
Dị hợp tử α+: α+/ α Thal.
Dị hợp tử α°, α+: α° / α+Thal.
β thalassemia
Gen chỉ đạo tổng hợp chuỗi β (gen β) nằm trên cánh ngắn nhiễm sắc thể 11 cùng các gen δ, γ, ε. Nếu nhiễm sắc thể (nhiễm sắc thể) tổn thương mất hoàn toàn khả năng chỉ đạo tổng hợp chuỗi p gọi là β°.
Nếu gen p tổn thương làm giảm tốc độ tổng hợp chuỗi β gọi là β +. Như vậy các kiểu tổ hợp:
Đồng hợp tử β° Thal: β°/ β°.
Dị hợp tử p° Thal: β°/ β.
Đồng hợp tử β +Thal: β+/ β+.
Dị hợp tử β+ Thal: β+/ β.
Dị hợp tử β°/ β +Thal: β°/ β+.
γ, δ thalassemia.
Không tổng hợp được chuỗi γ và δ: ít có ý nghĩa lâm sàng.
Phân bố địa lý
β Thal: Phân bố từ Địa Trung Hải, châu Phi qua Nam Ấn đến châu Á và Đông Nam Á, ở Việt Nam tỷ lệ mắc bệnh cao ở dân tộc ít người theo mức giảm dần từ vùng Bắc đến Trung đến Nam.
Α Thal: Gặp nhiều ở châu Á, nhất là Đông Nam Á. ở Việt Nam: tỷ lệ mắc bệnh cao ở dân tộc ít người theo thứ tự các vùng Trung - Nam - Bắc.
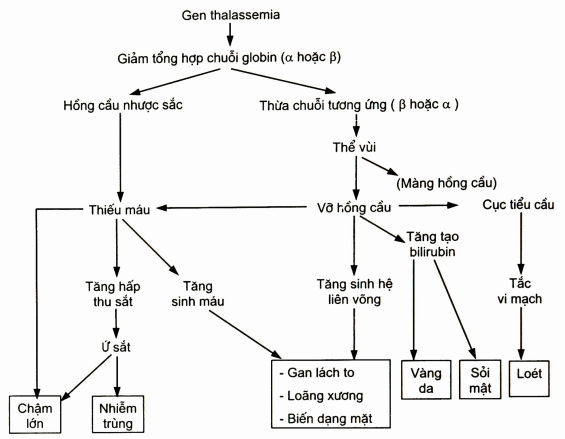
Sơ đồ. Cơ chế hình thành triệu chứng lâm sàng trong thalassemia
Cơ chế hình thành các biểu hiện lâm sàng
Khi gen globin bị tổn thương, chuỗi globin đó không được tổng hợp hay tổng hợp bị giảm nên thiếu huyết sắc tố gây thiếu máu. Nhưng căn nguyên gây bệnh chính là thừa tương đối chuỗi tương ứng. Các chuỗi globin thừa này sẽ trùng hợp tạo nên các thể vùi huyết sắc tố. Những thể vùi này không có tác dụng vận chuyển oxy và gắn lên màng hồng cầu làm thay đổi tính thấm, tính mềm dẻo của màng hồng cầu làm hồng cầu dễ vỡ. Hồng cầu vỡ gây thiếu máu và các biểu hiện lâm sàng.
α thalassemia
Là bệnh do giảm hoặc mất hẳn sự tổng hợp chuỗi α.
Cơ chế phân tử và sinh bệnh
Bình thường gen α nằm trên cánh ngắn nhiễm sắc thể 16, mỗi nhiễm sắc thể có hai gen α như vậy một cơ thể bình thường có bốn gen α.
α Thal do mất đoạn nhiễm sắc thể.
Tổn thương mất đoạn nhiễm sắc thể có thể mất một hoặc hai gen.
Trường hợp mất một gen còn gen kia vẫn hoạt động, đó là α Thal 2, còn gọi α+ thalassemia (α + Thal).
Trường hợp mất cả hai gen là α Thai I còn gọi là α° Thal.
α Thal không phải do mất đoạn nhiễm sắc thể.
Có nhiều đột biến điểm: thay thế, thểm hoặc mất một vài base nitơ sẽ làm giảm hoặc mất hẳn tổng hợp chuỗi a. Ví dụ, đột biến điểm ở codon (bộ ba) kết thúc từ TAA thành CAA, vì đột biến đó mà quá trình sao chép không dừng lại ở chỗ đáng dừng mà kéo dài, tạo ARNm không bền vững nên quá trình tổng hợp chuỗi α bị giảm hẳn (α Thal 2 hay α+ Thal).
Như vậy tùy theo số gen α bị mất mà tạo nên các tình trạng bệnh lý.
Biểu hiện lâm sàng
Như đã trình bày trên, có nhiều mức độ tổn thương các gen α (từ mất 1 đến mất 4 gen α). Do đó khả năng tổng hợp chuỗi a cũng giảm tùy theo các kiểu tổ hợp gen. Từ đó các bệnh cảnh lâm sàng có khác nhau. Các dạng thể hiện lâm sàng là:
Huyết sắc tố Bart’s (bệnh phù thai)
Cấu tạo phân tử là γ4:
Đây là dạng thalassemia nặng nhất, 100% chết trước hoặc ngay sau khi đẻ. Biểu hiện là thai phù, vàng da và thiếu máu, gan to, rau to và mủn, mẹ thường bị ngộ độc thai nghén khi mang thai. Xét nghiệm điện di huyết sắc tố thấy thành phần huyết sắc tố chủ yếu là Hb Bart’s.
Do ở bào thai bình thường huyết sắc tố chủ yếu là Hb F gồm 2 chuỗi α và 2 chuỗi γ. Khi mất cả 4 gen α cơ thể không tổng hợp được chuỗi α nên 4 chuỗi γ trùng hợp (γ4 tạo Hb Bart’s. Huyết sắc tố Bart’s có ái lực cao với oxy nên không thể nhả oxy cho tổ chức do vậy gây thiếu oxy và tử vong.
Bệnh được phát hiện khi thấy thai phù, chết lúc đẻ hoặc sẩy, nhất là vùng Đông Nam Á.
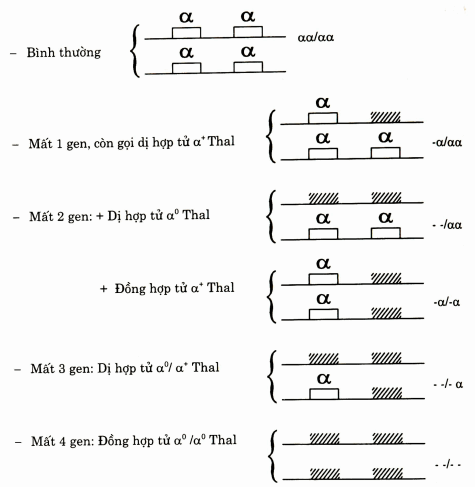
Hình. Các kiểu tổ hợp gen trong α thalassemia
Bệnh Hb H
Chỉ còn 1 gen α hoạt động (do mất đoạn hoặc đột biến làm ngừng tổng hợp ở 3 gen). Biểu hiện tan máu nhẹ có thể xuất hiện khi mới sinh (ít gặp), thường nhất là có các cơn tan máu nặng lên khi có đợt nhiễm trùng, xét nghiệm thấy hồng cầu nhỏ, nhược sắc, có thể Heinz (thể vùi).
Điện di huyết sắc tố có: Hb A, Hb H (khi mới sinh có Hb Bart’s).
α Thai thể nhẹ: mất hai gen α
Thường không có biểu hiện lâm sàng, xét nghiệm có thể thấy hồng cầu nhỏ, lúc mới sinh nếu điện di Hb có thể thấy Hb Bart’s (khoảng 2-5%).
α Thai thể ẩn: chỉ mất một gen α.
Không có triệu chứng lâm sàng, nếu xét nghiệm điện di Hb lúc trẻ mới sinh có thể thấy Hb Bart’s khoảng 1-2%.
β thalassemia
Là bệnh gây ra do giảm hoặc mất hẳn sự tổng hợp chuỗi β globin.
Cơ chế phân tử và sinh bệnh
Có rất nhiều khuyết tật ở gen β gây β Thai. Các đột biến chủ yếu là thay thế hay mất một vài gốc base, làm ảnh hưởng đến quá trình sao chép ARN, chín ARN và dịch mã.
Các đột biến ở vùng khởi động làm giảm tốc độ sao chép, gây β+ Thal.
Đột biên ở một số bộ ba mã hoá (codon) làm thành mã chấm hết, không tạo được ARNm đầy đủ gây β° Thal.
Các đột biến ở đoạn đầu sao chép hay đoạn cuối sao chép làm rối loạn quá trình sao chép ARNm, gây giảm tốic độ tổng hợp chuỗi β đó là β+ Thal.
Các đột biến ở vùng intron làm chậm quá trình chín của ARNm gây β+ Thai
Chuỗi β giảm hoặc không được tổng hợp sẽ làm cơ thể tăng cường tổng hợp chuỗi khác để bù:
Tổng hợp chuỗi δ, tạo α2δ2 đó là Hb A2.
Tổng hợp chuỗi γ, tạo α2γ2 đó là Hb F.
Các chuỗi α thừa ra lắng đọng vào màng hồng cầu gây vỡ hồng cầu và có các hậu quả bệnh lý.
Biểu hiện lâm sàng (các thể lâm sàng)
Tùy theo mức độ tổn thương của gen β (β° và các mức độ β+ Thal).
Tùy theo tổ hợp gen: (đồng hợp tử hay dị hợp tử) mà có các biểu hiện lâm sàng khác nhau.
Thể β thalassemia nặng (thiếu máu Cooley)
Là thể gây ra do đồng hợp tử β Thal.
Lâm sàng: biểu hiện rất sớm khi trẻ mới sinh được vài tháng đến vài tuổi (lúc mà đáng ra gen β hoạt động thay gen γ) thể hiện vàng da, gan có thể to, X quang xương có hình chân tóc, thiếu máu nặng.
Xét nghiệm:
+ Huyết đồ: hồng cầu nhỏ, nhược sắc, có mảnh hồng cầu, huyết sắc tố giảm nặng, hồng cầu biến dạng, tăng hồng cầu lưới.
+ Điện di Hb: HbA giảm hoặc không có, Hb A2 tăng (8-9%), Hb F tăng cao (20 - 99%).
Các kiểu gen: β°/β°; β°/β+; β+/β+.
Thể β Thal trung gian
Thường có biểu hiện thiếu máu muộn hơn thể nặng và bệnh cảnh lâm sàng nhẹ hơn, triệu chứng chính là thiếu máu.
Xét nghiệm:
Huyết đồ: hồng cầu nhỏ, nhược sắc, biên dạng, có hình răng cưa, tăng hồng cầu lưối.
Điện di HST: Hb A giảm, Hb A2 tăng (có thể đến 10%); Hb F tăng có thể
90 - 99%.
Kiểu tổ hợp gen β°/β; β+/β+; β+/β0; β7(αβ°).
Thể nhẹ
Biểu hiện nhẹ hoặc không có dấu hiệu lâm sàng.
Xét nghiệm:
Huyết học: tăng số lượng hồng cầu, hồng cầu nhỏ, nhược sắc.
Kiểu gen: β7β; β°/β; β+/β+.
Bài viết cùng chuyên mục
Huyết sắc tố bất thường (tổng hợp chuỗi globin bất thường)
Tùy theo sự thay đổi mà có các biểu hiện khác nhau, rất nhiều loại thay đổi được phát hiện, Tên các huyết sắc tố bất thường được đặt theo địa dư phát hiện nhưng có tên thông nhất.
Bệnh lupus ban đỏ hệ thống (Systemic Lupus Erythematosus - SLE)
Triệu chứng lâm sàng biểu hiện trên nhiều tạng trong cơ thể bệnh nhân, nhưng thường gặp nhiều hơn là sốt, triệu chứng ở da, triệu chứng cơ - khớp, triệu chứng về huyết học, thận và tim mạch.
Lơ xê mi kinh dòng lympho - Bệnh tăng sinh Lympho mạn ác tính
Người ta nhận thấy lơ xê mi kinh dòng lympho có nhiều ở châu Au và Mỹ, ít gặp hơn ở châu Á, Ở Mỹ có thể gặp với tỷ lệ cao 2 đến 3 người trên 100.000 dân
Bệnh Hemophilia
Bệnh hemophilia là bệnh dễ chảy máu (máu khó đông) do thiếu (hay bất thường) các yếu tố tạo thành thromboplastin nội sinh đó là các yếu tố VIII, IX hay XI .
Các xét nghiệm và ý nghĩa thực tiễn đánh giá sinh lý sinh hóa máu
Chuỗi phản ứng men tác động lên chuyển hóa acid arachidonic tạo ra nhiều chất gây tăng thấm mạch, đồng thời tác động lên hệ thống đông máu gây rối loạn đông máu, đông máu rải rác trong lòng mạch, chảy máu.
Bổ thể trong huyết học truyền máu
C8, C9 hai thành phần cuối cùng bị hoạt hoá sẽ tạo ra các lỗ thủng làm thay đổi tính thấm màng tế bào, làm tế bào trương to và chết.
Nguyên tắc và các bước thực hiện truyền máu lâm sàng
Nguyên tắc chỉ định truyền chế phẩm máu hiện nay trên thế giới, và ở Việt Nam, là chỉ định truyền máu hợp lý, trên cơ sở các biểu hiện lâm sàng, và xét nghiệm.
Bạch cầu, cytokin, chất trung gian và gốc tự do trong máu bảo quản
Trước hêt, do xuất hiện một số men bạch cầu làm pH máu bảo quản giảm, pH giảm gây nhiều bất lợi trong đó có một bất lợi đáng chú ý là tạo điều kiện hình thành các gốc tự do có nhiều tác hại.
Phân loại và chẩn đoán hội chứng tăng sinh Lympho mạn ác tính
Xét nghiệm miễn dịch: CD3, CD4, CD34, CD19, CD20, Xét nghiệm tế bào di truyền: biến đổi nhiễm sắc thể, Xét nghiệm kháng nguyên gây ung thư.
Phân loại và chẩn đoán suy tủy
Dịch hút tuỷ xương: tuỷ đồ, tương tự như máu ngoại vi, số lượng tế bào tuỷ giảm, Sinh thiết tuỷ nghèo tế bào, tổ chức mỡ lấn át (mỡ hoá tuỷ), xơ hoá, thâm nhiễm lympho.
Tăng tiểu cầu nguyên phát (primary thrombocytopenia)
Hoàn cảnh phát hiện: bệnh thường gặp ở người trên 50 tuổi được phát hiện tình cờ khi làm xét nghiêm hệ thống hoặc khi vào viện vì các biến chứng tắc mạch hoặc chảy máu.
Đa u tủy xương (Multiple Myeloma)
Bệnh với các biểu hiện bệnh lý: khuyết và loãng xương, giảm sinh tủy, tăng tương bào tại tủy xương, tăng độ nhớt máu, tăng protein đơn dòng, giảm chức năng thận hoặc suy thận.
Bệnh xuất huyết giảm tiểu cầu
Giảm tiểu cầu miễn dịch ở trẻ sơ sinh là do bất đồng kháng nguyên HPA giữa mẹ và con, gây miễn dịch ở mẹ và giảm tiểu cầu ở con. Hầu hết các trường hợp được chẩn đoán sau đẻ.
Bệnh lý suy giảm miễn dịch
Xuất hiện các kháng thể tự miễn dịch như kháng thể chống tiểu cầu, chống bạch cầu trung tính, kháng thể chống lympho, nhưng lại không phát hiện thấy kháng thể chống nhân và yếu tố dạng thấp.
Phân loại và chẩn đoán hội chứng rối loạn sinh tủy
Số lượng hồng cầu, bạch cầu, tiểu câu, huyết sắc tố. Hình thái các loại tế bào. Tuỷ Tuỷ đồ: tế bào học. Sinh thiết tuỷ: tổ chức tuỷ.
Những tiêu chuẩn cho máu an toàn
Đốì với nam không nên cho quá 4 lần trong một năm. Đối với nữ không nên cho quá 3 lần trong 1 năm. Đối với cho huyết tương, cho tiểu cầu thời gian quy định khác.
Giảm sinh tủy - Suy tủy xương (Aplastic anemia)
Phần lớn bệnh nhân Fanconi không đáp ứng với ATG hay cyclosporin A, nhưng có đáp ứng tốt vối androgen, Bệnh nhân tủ vong ở tuổi 10 đến 20 tuổi khi suy tủy ngày càng nặng.
Truyền máu tự thân và ứng dụng
Phải kiểm tra các thành phần bạch cầu, hồng cầu ly giải trong đơn vị máu, tuy nó không có nguy hại lớn về lâm sàng như truyền máu đồng loài, nhưng khi có hàm lượng cao thì truyền cho người bệnh cũng có thể xảy ra tai biến.
Quá trình sinh máu bình thường
Trong quá trình phát triển, tế bào nguồn sinh máu có khả năng sinh sản và biệt hoá thành các tế bào máu trưởng thành có chức năng riêng biệt, Người ta chia tế bào nguồn sinh máu thành bốn loại.
Kháng nguyên kháng thể trong huyết học truyền máu
Kháng nguyên không hoàn toàn là kháng nguyên chỉ có phần đặc hiệu mà không có phần mang tính kháng nguyên, nếu có một mình, chúng không gây đáp ứng miễn dịch.
Phân loại bệnh lý tế bào nguồn sinh máu và bệnh máu
Tuy phân làm hai nhóm, nhưng cả hai liên quan và gắn bó với nhau rất chặt chẽ như bệnh lý của tuỷ xương lại được phản ánh ở máu ngoại vi và số lượng và hình ảnh máu ngoại vi cũng phản ánh bệnh lý của tuỷ xương.
Điều hòa quá trình sinh máu
Ngược lại với các yếu tố phát triển, các yếu tố ức chế sinh máu có thể can thiệp vào một hoặc nhiều khâu khác nhau, một hay nhiều dòng tế bào, hạn chế quá trình tăng sinh, biệt hoá và hoặc chức năng của tế bào.
Nguồn gốc phát triển, cấu trúc và chức năng của tiểu cầu
Bình thường tiểu cầu không dính vào thành mạch, có lẽ do một chất có tác dụng ức chế dính của tiểu cầu - chất đó có thể là prostaglandin. Tuy nhiên khi có đứt mạch máu thì lập tức tiểu cầu được hoạt hóa và dính vào nơi tổn thương.
Tổng hợp huyết sắc tố (Hb)
Mỗi loại globin là sản phẩm của một gen, nên cũng có 2 họ gen globin (hình) đó là họ gen a và họ gen không α. Trong các nguyên hồng cầu, tổng hợp globin cũng qua các giai đoạn mã hoá, chín ARN, thông tin và phiên mã.
Phân loại và chẩn đoán Lơxêmi cấp
Tuỷ đồ, tế bào tăng sinh bất thường, tế bào blast trên 30 phần trăm trong số tế bào có nhân, Sinh thiết, tuỷ tràn ngập tế bào blast, các dòng khác bị chèn ép tổ chức tuỷ bị lấn át.
