- Trang chủ
- Sách y học
- Bài giảng huyết học và truyền máu
- Lơ xê mi kinh dòng lympho - Bệnh tăng sinh Lympho mạn ác tính
Lơ xê mi kinh dòng lympho - Bệnh tăng sinh Lympho mạn ác tính
Người ta nhận thấy lơ xê mi kinh dòng lympho có nhiều ở châu Au và Mỹ, ít gặp hơn ở châu Á, Ở Mỹ có thể gặp với tỷ lệ cao 2 đến 3 người trên 100.000 dân
Biên tập viên: Trần Tiến Phong
Đánh giá: Trần Trà My, Trần Phương Phương
Lơ xê mi kinh dòng lympho là bệnh ác tính mang đặc điểm của dòng lympho trưởng thành ở máu, ở tủy và ở tổ chức lympho. Ở Mỹ và các nước châu Âu tỷ lệ khá cao ở người có tuổi (> 50) khoảng 2,7 người bị trên 100.000 dân, chiếm 0,8% trong các bệnh ung thư và 30% của các lơxêmi. Bệnh thuộc dòng B lympho, nhưng có khoảng 2% thuộc T lympho. Bệnh hiếm gặp ở các nước châu Á, Việt Nam rất ít gặp.
Về lịch sử, bệnh được mô tả đầu tiên bởi Wirchow năm 1840, sau đó Ehrlich (1891) bàn tới phương pháp nhuộm hoá tổ chức để phát hiện bệnh, phương pháp này được Turk (1903) xác nhận là một kỹ thuật phát hiện lơ xê mi kinh dòng lympho và lymphosarcoma có hiệu quả, năm 1954 lần đầu tiên Tivey công bố tài liệu với 685 lơ xê mi kinh dòng lympho và thấy thời gian sống của họ trung bình là 3 năm. Đồng thời những nhà lâm sàng Ý thông báo một số thuốc có tác dụng điều trị lơ xê mi kinh dòng lympho như corticoid, các hoá chất thuộc nhóm alkyl. Tới 1967 Dameshek giả thiết rằng lơ xê mi kinh dòng lympho là bệnh của lympho không có khả năng miễn dịch, tiếp theo giả thuyết này, nhiều người chứng minh bằng Ig (Immunoglobulin) trên bề mặt tế bào và kết luận chúng là tế bào B lympho. Trong các năm 70, Rai và nhiều tác giả mô tả bệnh cảnh lâm sàng của bệnh bao gồm thiếu máu, giảm tiểu cầu - đồng thời các tác giả cũng đưa ra một số thuốc có hiệu quả kéo dài lui bệnh như tludarabin (2 chlorodeoxy adenosin, cladribin) và ứng dụng phương pháp ghép tủy...
Sinh bệnh học
Bệnh sinh học của bệnh còn nhiều bí ẩn, với nhiều ý kiến khác nhau.
Do yếu tố di truyền
Người ta nhận thấy lơ xê mi kinh dòng lympho có nhiều ở châu Au và Mỹ, ít gặp hơn ở châu Á. Ở Mỹ có thể gặp với tỷ lệ cao 2-3 người/100.000 dân. Lơ xê mi kinh dòng lympho có tính chất gia đình, tuy nhiên điều này đến nay chưa được khẳng định.
Do không bình thường gen di truyền
Về mặt này, có nhiều tài liệu thông báo, thường gặp dạng bất thường đơn dòng nhiễm sắc thể (NST) thí dụ có thể gặp biến đổi của nhiễm sắc thể 12 13 14.
Bất thường nhiễm sắc thể 12:
Trisomy 12 (17%).
12q+.
Nhiễm sắc thể 13: thường gặp các dạng sau:
13q, 13a14.
Gen RBl (Retinoblastoma) xuất hiện 13q+14.
Nhiễm sắc thể 14: bất thường nhiễm sắc thể 14 thuồng gặp dạng sau:
14q+11.
q32, t(11; 14) (q13;q32); t(14;19)(q32;q13).
BCL-1 (B-cell leukemia), BCL-2, BCL-3. Các gen này đóng vai trò bệnh lý gen trong lơxêmi cấp.
Nhìn chung các biến đổi nhiễm sắc thể hoặc biến đổi gen nói trên cũng chỉ là những hiện tượng phát hiện thấy. Bệnh lý gen chính thức của lơ xê mi kinh dòng lympho tới nay chưa được khẳng định. Tuy nhiên, nghiên cứu về gen cho thấy hiện tượng: bệnh lý T lympho thường xảy ra ở nhiễm sắc thể 14 (14qll), còn bệnh lý của B lympho lại xảy ra ở điểm gãy nhiễm sắc thể14 ở vị trí 3 (14q32).
Vai trò của các dấu ấn miễn dịch
Thường gặp các phenotyp dòng B lympho như CD5, CD19, CD20,CD37, CD39, rất hiếm gặp CDlla, CD54.
Các Ig bề mặt: đặc biệt chú ý chuỗi nhẹ Lambda hoặc Kappa. Người ta nhận thấy có tới 60% bệnh nhân lơ xê mi kinh dòng lympho có bất thường chuỗi nhẹ K, còn 40% ở chuỗi nhẹ Lambda, chuỗi nặng ít biến đổi, nếu có chỉ xảy ra ở vùng V (Varian).
Bất thường miễn dịch:
+ Tự miễn dịch: có thể phát triển kháng thể chống lại tế bào nguồn của từng dòng tế bào gây suy tủy: suy tủy dòng hồng cầu, suy dòng mẫu tiểu cầu, gây thiêu máu tan máu, gây giảm bạch cầu trung tính, trên lâm sàng thấy một số bệnh nhân thấp khớp trỏ thành lơ xê mi kinh dòng lympho.
+ Suy giảm miễn dịch: một phần do bệnh lý tế bào B lympho, phần khác do điều trị hoá chất. Trạng thái này gọi là giảm miễn dịch mắc phải.
+ Giảm y-globulin: bệnh nhân thường bị nhiễm trùng do giảm IgM và IgG.
+ Giảm miễn dịch tế bào: bệnh nhân bị lơ xê mi kinh dòng lympho giảm hoặc không phản ứng với PHA, giảm cytokin do giảm T-CD4 và T-CD8.
Đặc điểm lâm sàng và xét nghiệm
Lâm sàng
Chủ yếu gặp ở nam giới.
Tuổi: bệnh nhân thường ở người cao tuổi 50 - 90 tuổi, rất hiếm ở tuổi trẻ < 25.
Có khoảng 1/4 số bệnh nhân không có triệu chứng gì cho chẩn đoán.
80% số bệnh nhân được chẩn đoán hạch to, hạch to nhiều nơi, chắc, không đau, hạch riêng rẽ, không liên kết, đây là dấu hiệu có ích nhất cho chẩn đoán sớm.
Gan lách to: thường xuất hiện ở giai đoạn toàn phát, hoặc giai đoạn cuối - lách và gan to chiêm khoảng 50% số bệnh nhân vào viện.
Các dấu hiệu lâm sàng khác:
+ Thiếu máu: thiếu máu từ từ, bệnh nhân ít để ý.
+ Xuất huyết do giảm tiểu cầu.
+ Nhiễm trùng do giảm miễn dịch thể dịch và miễn dịch tế bào, giảm bạch cầu hạt trung tính, giảm miễn dịch tế bào làm bệnh nhân dễ bị nhiễm virus, nhiễm nấm.
Nhìn chung, các triệu chứng lâm sàng rất lu mờ, hạch to, gan lách to, bệnh nhân không chú ý, cho nên có 1/4 trường hợp phát hiện được bệnh là do bệnh nhân đi khám một bệnh khác.
Xét nghiệm
Máu:
Tăng lympho trưởng thành về số lượng tuyệt đối, có khi tăng trên 5 G/lít, thậm chí tăng > 200 G/lít. Trên tiêu bản máu đàn có tới 70-90% là các lympho nhỏ nhân đậm đặc, nguyên sinh chất hẹp.
Hình thái hồng cầu bình thường, số lượng hồng cầu giảm, tiểu cầu giảm.
Dòng bạch cầu hạt giảm nặng.
Tủy:
Tế bào lympho xâm lấn, lấn át toàn bộ tủy, các tổ chức mỡ bị lấn át.
Huyết thanh học về miễn dịch:
Các Ig (IgG, IgM, IgA) giảm nặng
Di truyền:
Biến đổi nhiễm sắc thể có thể gặp các dạng sau đây:
Trisomy nhiễm sắc thể 12, 14q.
Chuyển đoạn t(11; 14)
Phát hiện gen ung thư BCL-2, gen này có mặt trên nhiễm sắc thể 22, 2, chúng đảm nhiệm sản xuất chuỗi nhẹ Ig.
Tế bào miễn dịch:
Hầu hết phát hiện thấy CD19+, CD2CB- thuộc dòng B lympho.
Chẩn đoán
Chẩn đoán xác định
Các dấu hiệu lâm sàng: hạch, lách, gan to không có triệu chứng như sốt, đau.
Kết quả xét nghiệm máu: bạch cầu tăng rất cao, chủ yếu là lympho trưởng thành, lympho nhỏ (> 5G/lít).
Dấu ấn MD: CD19+, CD20+.
Chẩn đoán các thể thay đổi (varian)
Thể không triệu chứng, kéo dài trong nhiều năm (Rai: gđ:0)
Lơ xê mi kinh dòng lympho + tan máu: Coombs (+), thiếu máu, HCL tăng.
Tiền lơxêmi dòng lympho do lơ xê mi kinh dòng lympho chuyển thành.
Thể T- Lơ xê mi kinh dòng lympho: tế bào lympho lớn, có hạt, thường gặp ở người trẻ (hiếm gặp), CD3+, CD7+, CD2+.
Lơxêmi tế bào NK: Có CD16, CD56(+).
Chẩn đoán phân biệt
U lympho: hạch phát triển nhanh, sốt, ra mồ hôi.
Tiền lơxêmi dòng T lympho: tế bào to (10-15pm), chẩn đoán MD (CD3, CD2+) thường do virus HTLV gây nên.
Lơxêmi tế bào tóc (hairy cell leukemia): tế bào bạch cầu có lông (tóc) thuộc B lympho. Tế bào lớn, nguyên sinh chất rộng, CD19(+), CD20(+).
Lơxêmi tế bào NK: lympho lớn, NSC có hạt lớn thuộc dòng T lympho CD3+, CD16+, CD56+.
Lơxêmi tương bào: hình dạng tương bào, CD38(+).
Điều trị
Dựa theo phân loại của Rai (bảng)
Trừ giai đoạn “0”, còn các giai đoạn I, II, III, IV đểu cần được điều trị, đặc biệt giai đoạn II và III có tủy giảm sinh, tan máu và hạch to nhiều nơi.
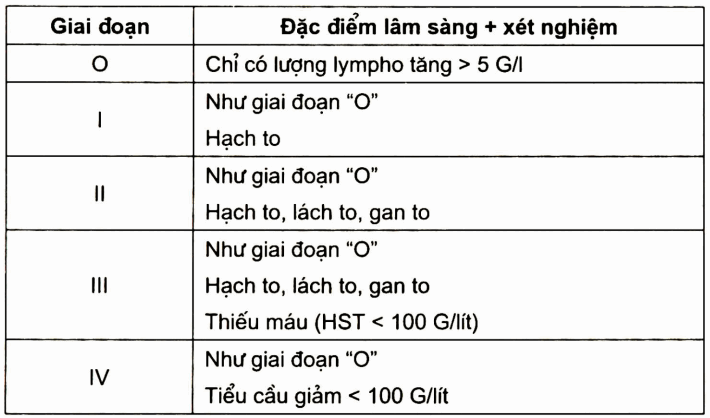
Bảng. Phân giai đoạn bệnh (Rai, 1997)
Điều trị đặc hiệu
Hoá chất
Corticoid:
30mg/m2/ngày, có thể dùng methylprednisolon: 1-2mg/kg/ngày x 5 ngày/tháng. Có thể phối hợp vối các chất thuộc nhóm alkyl như cyclophosphamid 250mg/m2/ngày x 5 ngày, có thể dùng liều cao 750mg/m2/ngày x 6 ngày rồi giảm dần 6 hoặc 4 mg/m2/ngày.
Phác đồ COP:
Cyclophosphamid: 250 mg/m2/ngày x 5 ngày, hoặc 750 mg/m2/ngày x 1 ngày tiêm tĩnh mạch.
Oncovin: 2 mg/m2/ngày x 1 ngày, tiêm tĩnh mạch.
Prednisolon: 40mg/m2/ngày x 4 ngày, mỗi tháng 1 liều như vậy, tiêm tĩnh mạch.
Phác đồ CHOP:
COP + Doxorubicin (hydroxodaunorubicin, adriamycin): 25 mg/m2/ngày, 1 tháng 1 lần.
Tia xạ
Thường dùng lOOc Gy/tuần X 10 tuần
Cắt lách
Khi lách quá to, cản trở hoạt động.
Điều trị hỗ trợ
Tăng cường miễn dịch:
+ y-globulin 4mg/kg/ngày X hàng tháng 1 lần.
+ Truyền lympho hòa hợp HLA.
Chống nấm, chống nhiễm trùng: kháng sinh.
Truyền tiểu cầu nếu tiểu cầu giảm < 50 G/lít + xuất huyết.
Yếu tố tiên lượng
Các yếu tố tiên lượng xấu:
Số lượng bạch cầu > 50 G/l.
Tiền lympho > 10%.
Thâm nhiễm tủy.
Tuổi >70
Đáp ứng chậm với hoá chất và tia xạ.
Một số thể lơ xê mi tế bào lympho hiếm gặp
Lơxêmi tế bào tóc (hairy cell leukemia)
Đặc điểm lâm sàng và xét nghiệm
Tuổi: gặp nhiều ở tuổi > 50
Giới: nam nhiều hơn nữ (4/1)
Lâm sàng: lách to (> 90%), thiếu máu, nhiễm trùng.
Biểu hiện của bệnh tự miễn dịch.
Cận lâm sàng:
+ Hồng cầu giảm nặng chiếm 2/3 các trường hợp.
+ Tế bào tóc ở máu tăng cao (> 90%), phản ứng với dấu ấn B lympho. Tế bào có hình dạng đặc biệt: bề mặt có nhiều “lông” kéo dài như sợi tóc, tế bào to (10- 15pm), nguyên sinh chất bắt màu kiềm nhạt, nhân có hạt nhân.
+ Tủy đồ, sinh thiết tủy: thâm nhiễm toàn bộ là tế bào tóc. Giảm sinh dòng hồng cầu, tiếu cầu, bạch cầu hạt. Có thể có tình trạng xơ tủy.
+ Hoá tế bào: tế bào kháng vói acid phosphatase.
Điều trị:
Nguyên tắc: chỉ điều trị khi có các điều kiện sau đây:
+ Lách to.
+ Thiếu máu (HST <100 g/lít).
+ Tiểu cầu <100 G/lít.
+ Bạch cầu hạt < 20 G/lít.
+ Thâm nhiễm tổ chức.
Thuốc:
+ Hoá trị liệu các hoá chất sau đây có thể dùng:
Chlorodeoxyadenosin (Cladribin): 0,09 mg/kg/ngày tiêm tĩnh mạch hoặc dưới da.
Deoxycoíbrmycin (Pentostatin): 4mg/m2/ngày, cách 1 tuần 1 lần, tiêm tĩnh mạch cho tới khi lui bệnh.
Fludarabin, chlorambucin, cyclophosphamid.
+ Interíeron (INF-a).
+ Cắt lách khi quá to.
+ Các chất kích thích sinh bạch cầu: G - CSF, GM-CSF.
Lơxêmi tế bào T (T-cell leukemia = TCL)
Bệnh lý phụ thuộc vào đột biến ở giai đoạn nào trong quá trình biệt hoá của T lympho, do vậy TCL có nhiều thể bệnh khác nhau. Thường là tế bào T chín (post thymic) vối các phenotyp CD3 (+), CD4(+), CD8(+), kích thước lớn, có thể chia 3 thể sau đây: Tiền T (Pro-TLL), T trưởng thành (Adult TLL) và NK -T lơxêmi (NK-T cells).
Tiền T-Lơxêmi (T-prolymphocytic leukemia = T - PLL)
Về lâm sàng: có thể gặp lách to, gan to, hạch to. Chậm hơn, có thể tổn thương da. Đây là triệu chứng có giá trị để phân biệt với lơ xê mi kinh dòng lympho và B-PLL.
Về xét nghiệm: cho thấy số lượng bạch cầu tăng 100-200G/lít, tế bào nhỏ, đều, CD4(+), CD8(-) chiếm 70%, CD4(+), CD8(+) chiếm 15%. Hầu hết tế bào này có CD7(+), đây là dấu ấn phân biệt với ATLL và NK-T. Sinh thiết các tổn thương da có thể thấy tế bào T thâm nhiễm tại chỗ.
Điều trị: gần giống ATLL (adult T-cell leukemia/lymphoma), TCL diễn biến nhanh, sống trung bình 6 tháng - phần lớn bệnh nhân không đáp ứng với hoá trị liệu. Theo kinh nghiệm của một số tác giả cho thấy có số ít bệnh nhân đáp ứng với phác đồ CHOP, số khác đáp ứng với DCF liều 4mg/m2 mỗi tuần, phác đồ này khoảng 10% lui bệnh hoàn toàn. Kết quả điều trị cho thấy nhóm bệnh nhân CD4(+) tốt hơn nhóm CD8(+). Nhìn chung TCL điều trị khó khăn tiên lượng xấu.
Lơxêmi-lympho T nguời trưởng thành (adult - T lymphocytic leukemia = ATLL)
ATLL liên quan HTLV do Gallo phát hiện 1977 và cũng năm này người ta đã mô tả lâm sàng và xét nghiệm của ATLL. Gen gây ATLL là HTLV-1, bệnh được mô tả đầu tiên ở Nhật, sau đó là ở Anh, Mỹ, Barazil.
Về lâm sàng: ít có các biểu hiện đặc biệt, ngoại trừ leukemia cấp. Có thể gặp: hạch to, tổn thương u da, tăng Ca++ máu, tăng lympho non > lOG/lít có khi tới 20-30 hoặc lOOG/lít, bệnh nhân có hội chứng lơxêmi cấp.
Chẩn đoán: dựa vào xét nghiệm có anti-HTLV-1 hoặc genom của HTLV bằng PCR. Các dấu ấn T lympho non như CD7(+), CD25(+) (anti-Tac). về hình thái ở máu và tủy thường thấy thể lympho non, nhân lớn, hình thái bất thường, số lượng bạch cầu tăng cao > 10 - lOOG/lít, hiếm có trường hợp > 100 G/lít.
Điều trị: bệnh nhân thường sống ngắn ngày (không quá 6 tháng). Một số ít bệnh nhân diễn biến chậm (dạng mạn tính) có thể sống > 1 năm. Điều trị bằng phác đồ CHOP hoặc M-BACOD có thể lui bệnh nhưng không duy trì được lâu. Cho tới nay điều trị bệnh này còn gặp nhiều khó khăn, tiên lượng xấu.
Lơxêmi - lympho hạt lớn (large granular lymphocytic leukemia = LGL)
Lympho lón có hạt trong nguyên sinh chất có dấu ấn dòng T lympho, đồng thời có dấu ấn tế bào NK (Natural Killer-cells). Do đặc điểm tế bào, bệnh này có các tên gọi khác nhau, vì có hạt azurophil trong nguyên sinh chất nên được gọi tên là lơxêmi lympho lớn có hạt (LGL). Vì bệnh kéo dài nên gọi là bệnh tăng sinh mạn dòng Tlympho (T-cell-CLL) hoặc lơxêmi dòng T - ức chế (T-suppressor cell - leukemia) hoặc NK-lơxêmi.
Về lâm sàng: có các biểu hiện chung của bệnh tăng sinh lympho kéo dài, dai dẳng, lách to, gan to, thiếu máu.
Xét nghiệm: bạch cầu tăng, chủ yếu là lympho, 20-70G/lít, giảm bạch cầu hạt trung tính, giảm hồng cầu, giảm HST. Có thể thấy tăng mẫu tiểu cầu, nhưng số lượng tiểu cầu ngoại vi giảm. Các dấu ấn màng (CD) cho thấy CD2,3 (+), CD4(-), CD8(+), CD16(+) trong một số ít trường hợp CD16(-) va CD4(+).
Chẩn đoán: dựa vào lâm sàng và các phenotyp của T lympho và NK như CD3(+), CD4, CD8, CD16...
Điều trị: hoá trị liệu bằng corticoid và chất thuộc nhóm alkyl như cyclophosphamid, chlorambucil hoặc methotrexat, trong một số nghiên cứu tác giả còn dùng cyclosporin A.
Nhìn chung bệnh có thể kéo dài, nhưng kém đáp ứng với điều trị, tiên lượng xấu.
Bài viết cùng chuyên mục
Giảm sinh tủy - Suy tủy xương (Aplastic anemia)
Phần lớn bệnh nhân Fanconi không đáp ứng với ATG hay cyclosporin A, nhưng có đáp ứng tốt vối androgen, Bệnh nhân tủ vong ở tuổi 10 đến 20 tuổi khi suy tủy ngày càng nặng.
Thiếu máu tan máu miễn dịch
Hiệu giá kháng thể miễn dịch rất thay đổi tăng lên rõ rệt ở môi trường albumin, còn hiệu giá kháng thể tự nhiên tăng lên rõ rệt ở môi trường muối.
Xơ tủy nguyên phát (myelofibrosis)
Nhìn chung ban đầu bệnh có biểu hiện là tăng sinh ở tủy xương với tăng số lượng tế bào, sau đó là giảm sinh tủy với giảm ba dòng tế bào máu ngoại vi.
Bệnh lý suy giảm miễn dịch
Xuất hiện các kháng thể tự miễn dịch như kháng thể chống tiểu cầu, chống bạch cầu trung tính, kháng thể chống lympho, nhưng lại không phát hiện thấy kháng thể chống nhân và yếu tố dạng thấp.
Lơ xê mi cấp - Ung thư máu cấp tính
Hiện nay, nguyên nhân gây bệnh lơ xê mi cấp vẫn chưa được xác định một cách chính xác. Yếu tố di truyền, thuốc, yếu tố môi trường, virus được đề cập đến như là những yếu tố nguy cơ gây bệnh.
Quá trình tăng sinh và biệt hóa các tế bào máu
Một tiền nguyên hồng cầu sinh ra hai nguyên hồng cầu ưa base I (erythroblast basophil) và thành bôn nguyên hồng cầu ưa base II. Tuy nhiên dưới kính hiển vi quang học, không thể phân biệt được nguyên hồng cầu ưa base I và nguyên hồng cầu ưa base II.
Chuyển hóa sắt và thiếu máu thiếu sắt
Ngoại trừ một số ít trường hợp quá tải sắt nặng, sắt tự do không có trong huyết tương do sắt được gắn với transferrin ở máu tĩnh mạch cửa.
Bổ thể trong huyết học truyền máu
C8, C9 hai thành phần cuối cùng bị hoạt hoá sẽ tạo ra các lỗ thủng làm thay đổi tính thấm màng tế bào, làm tế bào trương to và chết.
Hội chứng rối loạn sinh tủy (Myelodysplastic syndrome)
Có một số yếu tố được coi là yếu tố thuận lợi tham gia vào qúa trình sinh bệnh như tia xạ, hóa chất nhóm benzen, thuôc nhóm alkylan, virus.
Miễn dịch trung gian tế bào (cellular mediated immunity)
Các tế bào của các tổ chức và cơ quan bình thường có trên bề mặt kháng nguyên hệ HLA. Đó là những kháng nguyên gây đáp ứng miễn dịch tế bào.
Các cytokin và điều hòa tạo máu
Các cytokin có tác dụng điều hoà hoạt động và phát triển tế bào đích, do đó chúng đang được sử dụng trên lâm sàng để điều trị bệnh nhất là bệnh suy giảm miễn dịch.
Kỹ thuật phát hiện HLA và ứng dụng lâm sàng
Sử dụng xác định các antigen thuộc HLA-A, B, c, DR, DQ. Tuy nhiên HLA-DR kém nhậy đối với phản ứng độc tế bào, do vậy nhiều nước đã dùng kỹ thuật PCR để xác định kháng nguyên thuộc hệ HLA-DR.
Tăng tiểu cầu nguyên phát (primary thrombocytopenia)
Hoàn cảnh phát hiện: bệnh thường gặp ở người trên 50 tuổi được phát hiện tình cờ khi làm xét nghiêm hệ thống hoặc khi vào viện vì các biến chứng tắc mạch hoặc chảy máu.
Bệnh Hemophilia
Bệnh hemophilia là bệnh dễ chảy máu (máu khó đông) do thiếu (hay bất thường) các yếu tố tạo thành thromboplastin nội sinh đó là các yếu tố VIII, IX hay XI .
Kháng nguyên kháng thể trong huyết học truyền máu
Kháng nguyên không hoàn toàn là kháng nguyên chỉ có phần đặc hiệu mà không có phần mang tính kháng nguyên, nếu có một mình, chúng không gây đáp ứng miễn dịch.
Các thành phần của máu
Màng hồng cầu có kháng nguyên nhóm máu, các kháng nguyên này nằm trên bề mặt hồng cầu, chúng là các liên kết của carbohydrat lipid protein.
Hệ nhóm máu ABO, Rh, các hệ khác và an toàn truyền máu
Các kháng nguyên nhóm máu, là các sản phẩm protein trên màng hồng cầu, mà quá trình tổng hợp, những protein này được mã hóa.
Cơ chế đông máu cầm máu và các xét nghiệm thăm dò
Sự tiếp xúc của máu với tổ chức dập nát, sẽ phát động quá trình đông máu, chất có trách nhiệm là một lipoprotein gọi là yếu tố tổ chức.
Bệnh lupus ban đỏ hệ thống (Systemic Lupus Erythematosus - SLE)
Triệu chứng lâm sàng biểu hiện trên nhiều tạng trong cơ thể bệnh nhân, nhưng thường gặp nhiều hơn là sốt, triệu chứng ở da, triệu chứng cơ - khớp, triệu chứng về huyết học, thận và tim mạch.
Tổng hợp huyết sắc tố (Hb)
Mỗi loại globin là sản phẩm của một gen, nên cũng có 2 họ gen globin (hình) đó là họ gen a và họ gen không α. Trong các nguyên hồng cầu, tổng hợp globin cũng qua các giai đoạn mã hoá, chín ARN, thông tin và phiên mã.
Tăng đông và huyết khối
Một số tác giả gọi tăng đông là tình trạng tiền huyết khối. Tuy nhiên cần lưu ý răng huyêt khối thường ít khi do một nguyên nhân đơn độc gây nên mà thường là hậu quả của sự kết hợp của một số những rối loạn và yếu tố nguy cơ.
Các tiến bộ và hiệu quả truyền máu ở Việt Nam
Truyền máu phát triển ở hầu hết ở các bệnh viện trung ương, và bệnh viện tỉnh, truyền máu toàn phần chưa có chương trình quốc gia về an toàn truyền máu.
Nguồn gốc phát triển, cấu trúc và chức năng của tiểu cầu
Bình thường tiểu cầu không dính vào thành mạch, có lẽ do một chất có tác dụng ức chế dính của tiểu cầu - chất đó có thể là prostaglandin. Tuy nhiên khi có đứt mạch máu thì lập tức tiểu cầu được hoạt hóa và dính vào nơi tổn thương.
Lơ xê mi kinh dòng hạt
Lơ xê mi kinh dòng hạt là một bệnh ác tính hệ tạo máu, đặc trưng bởi sự tăng sinh các tế bào dòng bạch cầu hạt biệt hóa, hậu quả là số lượng bạch cầu tăng cao ở máu ngoại vi với đủ các tuổi của dòng bạch cầu hạt.
Ghép tủy tế bào nguồn
Ghép tuỷ tế bào nguồn, nghĩa là truyền tế bào nguồn vào máu như truyền máu, nhưng không dùng màng lọc máu, truyền chậm và dùng kim luồn vào tĩnh mạch trung tâm.
